SUTD and MIT scientists first to simulate a large-scale virus, M13
SUTD - Lunna Li and Desmond K. Loke
MIT - Angela M. Belcher
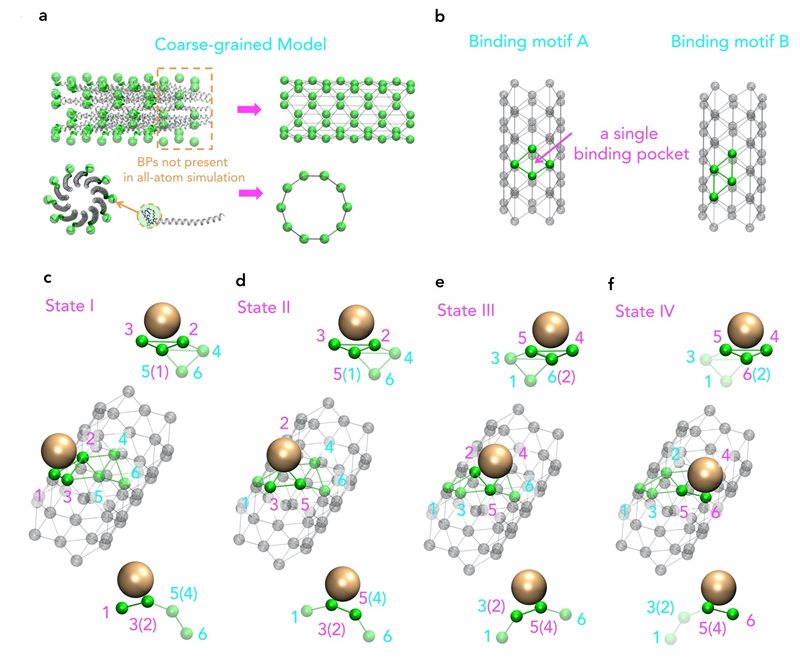 Molecular view of a coarse-grained model based on the original structure of M13 major coat proteins.
Molecular view of a coarse-grained model based on the original structure of M13 major coat proteins.
Atomistic simulations are a powerful tool to study the movement and interactions of atoms and molecules. In many biological processes, large-scale effects, for example, assembly of large viruses to nanoparticles are important. The assembly processes of these large viruses are of fundamental importance to the design of many devices and viral protein-targeted therapeutics. However, the time and length scale of these assembly processes are usually too large for simulations at molecular resolution.
Moreover, even though an increase in computing power allows for more complex and longer simulations, virus structures, such as M13, are still beyond reach. That is why a research group from the Singapore University of Technology and Design (SUTD) and the Massachusetts Institute of Technology (MIT) has developed a procedure that links large-scale assembly processes to molecular simulations. Assistant Professor Desmond Loke from SUTD’s Science, Mathematics and Technology cluster said, “For the simulation of M13, we started with different sets of force fields. Suitable force fields were chosen and they were used as the inputs for a molecule dynamics simulations with the coarse-grain model designed to capture key pattern of the assembly process.”
“While we know that M13-based manufacturing can be fundamentally driven by nanoparticle-peptide interactions, which may also be a key principle behind M13-type bioengineering, we have little knowledge of how repeated patterns of short-end-peptides on a M13 surface are actually involved in these interactions. To study this, we ideally have to include a full structure of the viral coat-protein, which is a difficult task for current state-of-the-art molecular dynamics simulations,” adds Dr Lunna Li, first author of the article.
The procedure allows users to add different types of nanoparticles to a solution, at a realistic level. Inspired by this procedure, Assistant Professor Loke and his colleagues were able to simulate a large-scale virus with nanoparticles and inside a solution for fifty nanoseconds.
Dr Li said, “The virus structure and solution contain about 700,000 atoms overall.” Considering the shape and size of the features, the complexity of this simulation can be larger than any simulation performed previously.
“A simulation performed in microseconds would have been possible if a smaller M13 model was used, but it can be worthwhile to reduce the time to actually observe how the full structure may influence the assembly between the M13 and nanoparticles,” explained Assistant Professor Loke.
MIT Biological Engineering Professor Angela Belcher, was also part of the research team that was simulating M13. This research was published in journal Nanoscale.
Reference:
Simulating selective binding of a biological template to a nanoscale architecture: a core concept of a clamp-based binding-pocket-favored N-terminal-domain assembly, Nanoscale. (DOI: 10.1039/D0NR07320B)
Acknowledgements:
We thank D. Frenkel, S. R. Elliott, G. J. Clausen, N. Heldman and T. H. Lee for important discussions. The authors acknowledge support from the Singapore University of Technology and Design (SUTDT12017003), Changi General Hospital (Singapore) (CGH-SUTD-HTIF2019-001), Ministry of Education (Singapore) (MOE2017-T2-2-064) and SUTD-Zhejiang-University (SUTD-ZJU (VP) 201903) grant programs. D. K. L. acknowledges support from the Massachusetts Institute of Technology–SUTD International Design Centre and National Supercomputing Centre, Singapore (15001618). L. L. acknowledges support from the MIT–SUTD Postdoctoral Fellowship.